
🚮 Más allá del contenedor de basura: Por qué tus lisosomas son la clave de la juventud y la enfermedad
- DRA. DYANA BUNNY

- 9 dic 2025
- 5 Min. de lectura
¿Alguna vez has visto esas películas apocalípticas donde los servicios públicos colapsan y la basura se acumula en las calles hasta que la ciudad se vuelve inhabitable? Bueno, eso es exactamente lo que le pasaría a tus células si no tuvieran a sus pequeños héroes anónimos: los lisosomas.
Durante décadas, en las clases de biología nos dijeron que el lisosoma era simplemente el "estómago" de la célula o su camión de basura. Pero hoy, como científica, vengo a decirte que nos quedamos cortos. El lisosoma no es un simple vertedero; es un centro de mando metabólico, una planta de reciclaje de alta tecnología y el secreto mejor guardado del antienvejecimiento.
Hoy vamos a sumergirnos en el ácido (literalmente) para entender cómo funciona, qué pasa cuando falla y por qué científicos como Christian de Duve y Yoshinori Ohsumi ganaron el Premio Nobel gracias a él.

Dr. Christian de Duve en su laboratorio
1. El Descubrimiento: Un accidente feliz en el laboratorio
La ciencia, a veces, es pura serendipia. En los años 50, el bioquímico Christian de Duve no estaba buscando un nuevo organelo; estaba estudiando una enzima del hígado. Pero notó algo raro: la actividad enzimática era baja en muestras frescas, pero se disparaba si la muestra se "dañaba" o envejecía. Lo llamó "latencia".
De Duve dedujo algo brillante: las enzimas destructoras estaban encerradas en "bolsas" de membrana. Si la bolsa se rompía, las enzimas salían a atacar. En 1955 bautizó a estas bolsas como lisosomas (del griego lysis, disolución, y soma, cuerpo). Básicamente, descubrió las granadas de mano de la célula.
2. Bioquímica Pura: Un baño de ácido controlado
Si has visto Alien, sabes que su sangre ácida derrite el suelo de la nave. El interior de un lisosoma es un poco así.
Para que las 60 hidrolasas ácidas (las tijeras moleculares que cortan proteínas, grasas y azúcares) funcionen, el interior del lisosoma debe ser muy ácido, con un pH entre 4.5 y 5.5. ¿Cómo logra la célula mantener esa acidez sin derretirse a sí misma?
La Bomba de Protones (v-ATPasa): Imagina una bomba de agua industrial que, en lugar de agua, empuja protones (H+) hacia adentro del lisosoma usando energía (ATP). Esto acidifica el ambiente.
El Campo de Fuerza (Glicocáliz): La membrana del lisosoma está recubierta internamente por una capa densa de azúcares (glicocáliz) que evita que las enzimas se coman la propia pared del lisosoma. Es como tener un estómago forrado de teflón ultra resistente.

3. Autofagia: El arte de "Marie Kondo" celular
Aquí es donde la cosa se pone de moda. Seguro has escuchado sobre el ayuno intermitente y la autofagia. Pero, ¿qué es realmente?
La autofagia es el proceso de reciclaje celular. Cuando la célula detecta que hay componentes viejos o dañados (como mitocondrias que ya no sirven), los envuelve en una bolsa de doble membrana llamada autofagosoma y los lleva al lisosoma para desmantelarlos.
Es como si Marie Kondo entrara a tu célula, tomara una mitocondria vieja y dijera: "Esto no nos da alegría (ni energía)", y la enviara al lisosoma para fundirla y crear piezas nuevas.
Este sistema es controlado por un interruptor maestro llamado mTORC1.
Con comida: mTORC1 dice "¡A crecer!" y bloquea el reciclaje.
En ayuno: mTORC1 se apaga, y se activan los factores TFEB/MiT que ordenan: "¡A limpiar la casa!". Se fabrican más lisosomas y se recicla todo lo viejo para obtener energía

Lamentablemente, con la edad, nuestra capacidad de hacer autofagia disminuye. Esto lleva a que acumulemos "basura" celular, lo que está detrás de enfermedades como el Alzheimer y el envejecimiento prematuro.
4. Cuando la fábrica falla: Enfermedades de Depósito Lisosomal (EALs)
Ahora, imagina que en esta planta de reciclaje perfecta, uno de los trabajadores se pone en huelga. Digamos, el encargado de triturar neumáticos. ¿Qué pasa? Que la planta se llena de neumáticos hasta el techo hasta que colapsa.
Esto es exactamente lo que ocurre en las Enfermedades de Depósito Lisosomal. Son enfermedades genéticas donde falta una sola enzima o proteína, provocando que un sustrato específico se acumule y "toxi-fique" la célula.
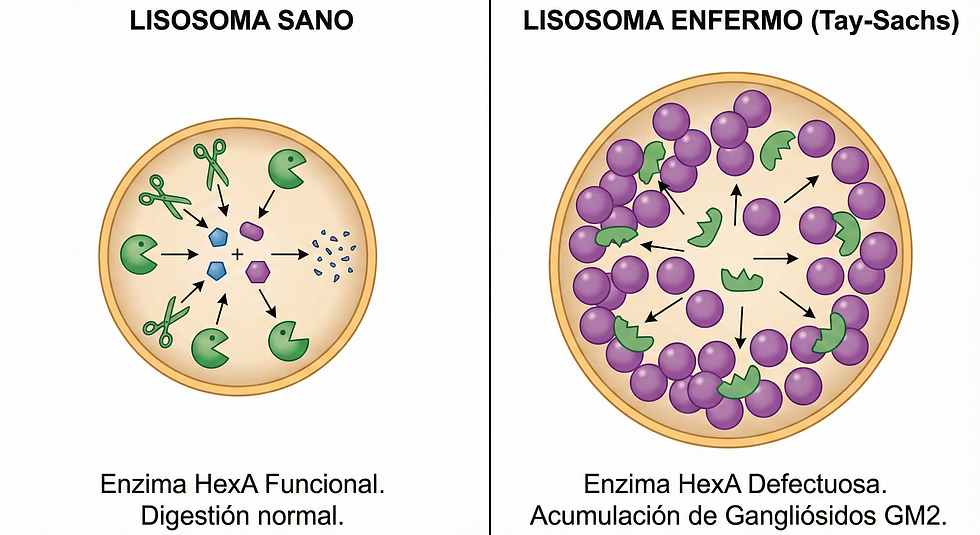
Aquí te presento a los "villanos" más conocidos de esta historia:
A. Enfermedad de Tay-Sachs
El fallo: Falta la enzima Hexosaminidasa A (HexA).
La basura: Se acumula un lípido llamado gangliósido GM2 en las neuronas.
El resultado: El cerebro se inflama y las neuronas mueren. Un signo clínico clásico es la "mancha rojo cereza" en el ojo del paciente. Es una enfermedad devastadora en niños.
B. Enfermedad de Gaucher
El fallo: Falta la enzima Glucocerebrosidasa.
La basura: Glucosilceramida.
El resultado: Los macrófagos (células de defensa) se llenan de grasa y parecen "papel arrugado" bajo el microscopio. Se acumulan en el bazo y el hígado, hinchándolos masivamente.
Ojo al dato: ¡Las mutaciones en este gen son un factor de riesgo importante para desarrollar Parkinson!.
C. Enfermedad de Pompe
El fallo: Falta la enzima que rompe el glucógeno (la reserva de azúcar).
El resultado: El glucógeno se acumula en los músculos, haciéndolos débiles. En bebés, el corazón se agranda enormemente (cardiomiopatía) porque no puede bombear bien.
D. Niemann-Pick Tipo C
El fallo: Aquí no falta una enzima que "corte", sino una proteína de tráfico (NPC1).
El resultado: Es un embotellamiento de tráfico. El colesterol entra al lisosoma pero no puede salir. La célula se llena de colesterol y lípidos, causando un daño neurodegenerativo severo.

5. El Futuro: ¿Podemos arreglar la maquinaria?
La ciencia no se ha quedado de brazos cruzados. Hemos pasado de solo observar a tratar activamente estas condiciones:
Terapia de Reemplazo Enzimático (TRE): Si falta la enzima, ¡se la ponemos! Inyectamos la enzima fabricada en laboratorio para que la célula la capture. Ha sido un milagro para pacientes con Gaucher y Pompe. Pero tiene un problema: a las enzimas les cuesta mucho entrar al cerebro (la barrera hematoencefálica es muy estricta).
Chaperonas Farmacológicas: Son moléculas pequeñas que actúan como "entrenadores personales" para las enzimas mutantes. Ayudan a que la enzima defectuosa se pliegue bien y llegue al lisosoma para trabajar.
Terapia Génica: La frontera final. Usar virus modificados (como taxis moleculares) para entregar una copia sana del gen defectuoso directamente a las células del paciente. Ya se está usando con éxito en algunas leucodistrofias y se investiga para Tay-Sachs.
Conclusión
Tus lisosomas no son simples bolsas de basura. Son guardianes sofisticados que deciden si tu célula crece, se repara o muere. Entenderlos no solo nos ayuda a curar enfermedades raras, sino que nos da las claves para entender el envejecimiento y enfermedades comunes como el Parkinson.
Así que la próxima vez que hagas ayuno o ejercicio, recuerda: estás entrenando a tus lisosomas para que te mantengan joven.



Comentarios